|
Abstract
Emerging evidence supports the pivotal role of renal
microvascular(Renal microcirculation, kidney microcirculation) disease as a determinant of tubulo-interstitial
and glomerular fibrosis in chronic kidney disease. An intact
microcirculation is vital to restore blood flow to the injured
tissues, which is a crucial step to achieve a successful repair
response. The purpose of this review is to discuss the impact
and mechanisms of the functional and structural changes of the
renal microvascular(Renal microcirculation,kidney microcirculation) network, as well as the role of these
changes in the progression and irreversibility of renal injury.
Damage of the renal microcirculation
(kidney microcirculation)and
deterioration of the angiogenic response may constitute early
steps in the complex pathways involved in progressive renal
injury. There is limited but provocative evidence that
stimulation of vascular proliferation and repair may stabilize
renal function and slow the progression of renal disease. The
feasibility of novel potential therapeutic interventions for
stabilizing the renal microvasculature(Renal microcirculation,kidney microcirculation) is also discussed. Targeted interventions
to enhance endogenous renoprotective mechanisms focused on the
microcirculation, such as cell-based therapy or the use of
angiogenic cytokines have shown promising results in some
experimental and clinical settings.
END-STAGE RENAL DISEASE (ESRD) is a growing health problem in
the adult U.S. population that consumed $23.9 billion in 2009, a
cost that has almost doubled in the past 10 years (55a). The
2009 United States Renal Data System(Renal microcirculation,kidney microcirculation) report shows that the
presence of chronic kidney disease (CKD) in current Medicare
patients reaches up to 16.2%, with an incidence of 5.8% per year
depending on the ethnicity (55a). Although hypertension and
diabetes are still the most common etiologies, the role of
vascular nephropathies and renovascular disease (RVD), a
progressive condition caused by narrowing of the renal arteries,
as causes of CKD and ESRD is on the rise among the elderly
population.
One of the main causes of chronic RVD is renal Renal microcirculation,kidney microcirculation()artery stenosis,
affecting 18¨C40% of those patients older than 65 (26) and almost
70% of patients with coronary or peripheral atherosclerotic
vascular disease (41). Mainly because of atherosclerosis (61),
RVD has a prevalence that ranges from 6.8% (32) to 23% (22), and
up to 15% of those patients will develop progressive
deterioration of renal function that may eventuate in CKD or
ESRD (26, 32).
A defective renal microcirculation(Renal microcirculation,kidney microcirculation), also known as microvascular (MV) disease
is a prominent pathological feature in CKD, irrespective of the
cause, and progresses as CKD evolves (30). In general, the
microcirculation is constituted by those vessels between 0 and
200 ¦Ěm, which are embedded within organs and are responsible for
the distribution of blood within tissues. Those small vessels in
the kidney include interlobar, arcuate, and interlobular
arteries, and smaller branching order microvessels like
arterioles, capillaries, and venules. Partly mediated by
augmented vasoconstriction and endothelial dysfunction in CKD
(78), MV disease can alter renal (Renal microcirculation,kidney microcirculation)blood flow and lead to a
progressive decrease in peritubular capillary flow and
consequently, mild tubulo-interstitial ischemia (63). Renal
ischemia could be observed both as a cause and a consequence of
damage in the kidney exposed to chronic RVD (46). An ischemic
insult is a powerful stimulus to trigger renal
neovascularization, a major physiological response that involves
a sequence of events resulting in development of new vessels
from preexisting ones. Vascular proliferation in the kidney not
only occurs during the developmental stages of the organ, but is
also a crucial mechanism by which the kidney faces a diversity
of insults (8, 36, 72, 77). Nevertheless, RVD is characterized
by intrarenal MV abnormalities that possibly aggravate the
effects of the vascular obstruction in the main renal(Renal microcirculation,kidney microcirculation) artery and
exacerbate the progression of renal (Renal microcirculation,kidney microcirculation)injury (6, 13, 49). It is
possible that the severity and persistence of damage in the
intra-renal MV bed may explain why the function of the stenotic
kidney is not always restored (only 30% of reperfusions are
successful) and sometimes even continues to deteriorate after
renal revascularization, the most frequent therapeutic approach
in patients with chronic RVD.
The purpose of this review is to discuss the impact of the
functional and structural changes of the renal microcirculation
(kidney microcirculation)and the role that such changes may play in the progression and
often irreversibility of renal injury in chronic RVD. I will
also discuss the potential mechanisms of renal MV injury in the
stenotic kidney and the feasibility of potential targeted
therapeutic interventions.
MV Disease and Renal Responses to Revascularization:
the Missing Link?
Restoration of tissue blood flow to
the site of ischemic injury is crucial for developing a
successful repair response. One of the most frequent therapeutic
approaches to treat the chronically stenotic kidney in humans is
by attempting restoration of blood flow either by opening the
stenotic renal(Renal microcirculation,kidney microcirculation) artery via renal angioplasty or through bypass
surgery. While the renal vascular obstruction may represent a
relatively straightforward problem to be addressed by
revascularization, the optimal therapeutic approach to treat RVD
is still not defined. Indeed, there is a significant gap between
the technical success of revascularization (achieved in almost
100% of the cases) (73) and the resulting improvements in renal
(Renal microcirculation,kidney microcirculation)function or resolution of hypertension (25¨C30%) (66, 67). The
severity of renal artery stenosis and the degree of impairment
of renal function at the time of reperfusion may predict the
relatively poor responses to revascularization (19, 20), but the
mechanisms behind the progressive deterioration of renal
function despite successful correction of the renal vascular
obstruction are still unclear. Since generation of new vessels
in the kidney seems to be a mechanism to preserve renal function
activated in response to injury (14, 36, 76), it is possible
that the damage of the renal (Renal microcirculation,kidney microcirculation)MV architecture and deterioration
of the angiogenic response constitute the early steps that lead
to progressive renal injury. The extent, severity, and
progression of MV damage in the ischemic renal parenchyma may
play a pivotal role in determining the success of
revascularization and explain why the ischemic kidney sometimes
does not improve or continues to deteriorate even after
restoration of blood flow. The factors determining these
relatively poor outcomes, however, are still unknown, and
advances in this field to understand the underlying mechanisms
of irreversible renal(Renal microcirculation,kidney microcirculation) injury are in dire need.
Potential Mechanisms Underlying Renal MV Disease
MV networks have the ability to adapt
to the local metabolic tissue requirements by upregulating or
downregulating angiogenesis, a process known as MV plasticity
(50). These responses involve the recruitment and activation of
numerous factors intertwined in a complex mechanism that
ultimately preserve the MV architecture and function. We will
briefly discuss them, as well as their role in inducing MV
disease.
Ischemia: role of vascular endothelial growth factor.
Thinking of ischemia as a key player
in renal (Renal microcirculation,kidney microcirculation)MV injury in RVD seems obvious. Although renal oxygen
supply is one of the highest in the body, and only 10% of
delivered oxygen is needed for metabolic demands, experimental
evidence has shown that renal artery stenosis can decrease
cortical and medullary oxygenation in the stenotic kidney (40,
58, 70). Facing a decrease in blood supply, the expected
physiological response to ischemia, as may occur in chronic RVD,
could include generation of new vessels to sustain tissue
perfusion. A key player in MV proliferation and repair is
vascular endothelial growth factor (VEGF), which shows different
patterns of expression and availability depending on the
ischemic environment. VEGF is crucial for preserving the
microvasculature, in general, and operates in concert with other
factors to promote cell division, migration, endothelial cell
survival, and tube formation that ultimately generate, repair,
and maintain MV networks, including those in the kidney.
VEGF increases significantly in cells exposed to acute hypoxia
(55), but eventually decreases when hypoxia is prolonged (57),
suggesting a biphasic regulation of VEGF and implying that cells
releasing VEGF themselves are injured or unable to secrete this
cytokine. In vivo experimental and clinical evidence correlates
with the in vitro studies, showing that chronic reductions of
renal (Renal microcirculation,kidney microcirculation)blood flow (14, 37), as occurs in RVD, progressive
glomerulopathies (60), and CKD (30), are associated with
significant reduction in VEGF. A recent clinical study
underscores the progressive nature of renal(Renal microcirculation,kidney microcirculation) MV disease in CKD,
revealing marked deficiencies in VEGF in these patients
accompanied by defective vascular repair, impaired angiogenic
responses, and enhanced vascular injury (30). We have previously
shown that chronic experimental RVD induces renal MV rarefaction
and decreases the expression and availability of renal (Renal microcirculation,kidney microcirculation)VEGF(14,
37, 76). Interestingly, these are also accompanied by decreased
expression of downstream mediators of VEGF, such as
angiopoietin-1 and Akt, suggesting a downregulation of the
angiogenic cascade in the stenotic kidney (Fig. 1). The
mechanisms for VEGF reduction in the stenotic kidney are not
entirely clear, but they appear to be a combined result of
altered post-transcriptional mechanisms (37) and a progressive
loss of the sources (24, 56) of this angiogenic cytokine. The
importance of VEGF for protecting the renal vasculature is
further underscored by studies from our laboratory (7, 37) and
others (42, 43), showing that intrarenal administration of VEGF
in RVD preserved the renal MV architecture and function,
improved renal blood flow and filtration function, and decreased
renal fibrosis. More importantly, we have also shown that
preserving the structure of the intra renal microcirculation
(kidney microcirculation)by
this intervention is functionally consequential, since it
significantly improved the renal responses to revascularization
by renal angioplasty (7), supporting the key role of MV disease
in determining the fate of the ischemic kidney.
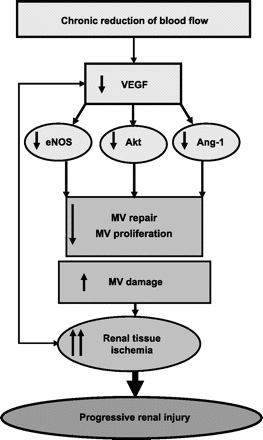 Fig.
1. Fig.
1.
Schematic illustration of the mechanisms leading to a decrease
in renal vascular endothelial growth factor (VEGF) and mediators
after a chronic reduction in blood flow, and the effects on the
renal microcirculation(kidney microcirculation). eNOS, endothelial nitric oxide synthase;
Ang-1, angiopoietin-1.
Inflammation
Inflammation is a prominent injurious mechanism activated in
CKD. Chronic inflammation has classically been shown in
pathological situations, such as rheumatoid arthritis, diabetes,
and cancer (23, 47), which also display increased angiogenesis
in an exacerbated manner. Inflammatory cells can directly
release angiogenic factors, such as VEGF, basic fibroblast
growth factor, and tumor-necrosis factor (TNF)-¦Á, among many
others, at inflammatory loci, which put forth mitogenic and
migratory effects in the endothelium, eventually promoting
vascular proliferation (23).
We have recently shown that cardiovascular risk factors, such as
lipid abnormalities (8, 12) and obesity (36), which are also key
risk factors for renal (Renal microcirculation,kidney microcirculation)disease, are associated with increased
renal expression of TNF-¦Á and significant renal cellular
inflammatory infiltrates. TNF-¦Á is a pivotal proinflammatory
mediator that also stimulates vascular proliferation directly
and by close interactions with other angiogenic cytokines, such
as VEGF, nuclear factor-¦ĘB, and interleukins (23). The increased
renal inflammation in these studies was accompanied by augmented
renal (Renal microcirculation,kidney microcirculation)MV density, likely reflecting a compensatory mechanism
that sustained renal function in the early stages of both
diseases. This concept was confirmed by arresting TNF-¦Á-induced
angiogenesis (8), which diminished MV density and decreased
renal blood flow and filtration function. Hence,
inflammation-induced angiogenesis seems to be an early but
partially protective compensatory mechanism, since the degree of
renal injury remained unchanged [reflected by the significant
tubulo-interstitial and glomerular damage in these models (8,
12, 36)] and likely progressive as the disease evolves. The
notion that these newly generated vessels in chronic
inflammatory milieus may promote the progression of tissue
damage is observed in the kidney and also in other diseases as
well (21, 45, 64). Inflammatory induced angiogenesis may indeed
result in highly permeable, leaky neovessels that allow
injurious cytokines to migrate to the extracellular space, thus
promoting tissue injury (Fig. 2).
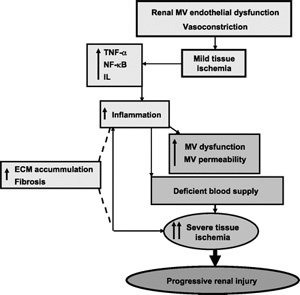 Fig.
2. Fig.
2.
Potential mechanisms and role of inflammation in promoting
microvascular (MV) dysfunction and injury in the stenotic
kidney. ECM, extracellular matrix; TNF, tumor necrosis factor;
NF-¦ĘB: nuclear factor-¦ĘB; IL, interleukin.
Renal (Renal microcirculation,kidney microcirculation)inflammation is also increased in experimental RVD (10,
11) and could trigger MV proliferation in the stenotic kidney.
Clinical and experimental studies have shown that the decline in
renal oxygenation precedes inflammation, fibrotic changes in
tubulo-interstitial cells, and matrix accumulation, suggesting
that hypoxia may both initiate and promote the renal tissue
damage (29). A chronic, sustained decrease in blood and oxygen
supply in the stenotic kidney may then activate hypoxia-induced
factors (36, 38), which, in turn, can promote inflammation and
angiogenesis (38). However, despite renal (Renal microcirculation,kidney microcirculation)inflammation, the stenotic kidney shows a marked reduction in MV density. This
does not rule out the deleterious role of inflammation in
aggravating renal injury and contributing to the progressive
fibrosis and deterioration of renal function in the chronically
stenotic kidney (10, 11). Since MV density is significantly
diminished in the stenotic kidney, the chronic ischemic insult
may surpass the proangiogenic phase of inflammation in the
stenotic kidney, possibly activating antiangiogenic mediators
(68) that accelerate the progression of renal(Renal microcirculation,kidney microcirculation) MV rarefaction. It
is possible that chronic hypoxia may potentiate inflammation and
further contribute to extracellular matrix accumulation and
increase renal fibrosis, in the absence of vascular regeneration
and combined with MV dysfunction as occurs in the stenotic
kidney.
Fibrosis
Irrespective of the etiology, renal fibrosis is the common final
stage of progressive renal disease. Pivotal renal profibrotic
factors often involved in chronic renal disease, such as
transforming growth factor-¦Â (28, 74) or connective tissue
growth factor (5), have potent effects in stimulating
angiogenesis. However, although we have shown that such factors
are also upregulated in the stenotic kidney (10, 11, 15), their
increase is accompanied not only by marked fibrosis, but also a
significant MV rarefaction. The accumulation of extracellular
matrix in the fibrotic kidney not only represents a buildup of
scar tissue, but also generates an active source of potential
antiangiogenic mediators, such as angiostatin, a potent
inhibitor of VEGF and downstream mediators (65, 75). Angiostatin
has been shown to be persistently elevated after ischemic renal
(Renal microcirculation,kidney microcirculation)injury and can significantly reduce VEGF-induced proliferation
and repair of peritubular capillaries, hence accelerating
tubular and interstitial damage (52). Other potent extracellular
anti-angiogenic factors and inhibitors of cell proliferation
that are highly expressed in kidneys are the thrombospondins
(35), which we have shown to be augmented in the atherosclerotic
kidney (9). In addition, we have previously shown that key
enzymes for matrix degradation and removal and MV development,
such as the matrix metalloproteinases-2 and -9, as well as the
antifibrotic and proangiogenic hepatocyte growth factor, are
reduced in the RVD kidney (11, 15). Decreased expression and
activity of these factors lead to further accumulation of
extracellular matrix, facilitating the buildup of intrarenal
fibrosis and feeding a vicious circle (Fig. 3). In addition, the
development of renal (Renal microcirculation,kidney microcirculation)scarring involved progressive changes and
likely loss of podocytes (27, 54), one of the main source of
VEGF (24), thus, in turn, further contributing to this
deleterious mechanism.
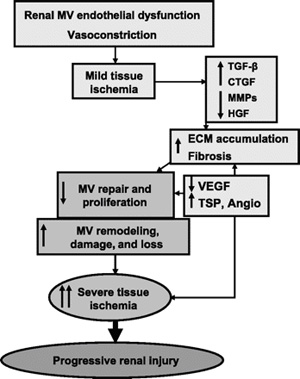
Fig. 3.
Schematic illustration describing the potential mechanisms and
role of renal(Renal microcirculation,kidney microcirculation) scarring in decreasing MV proliferation and
promoting MV remodeling, damage, and loss in the stenotic
kidney. TGF, transforming growth factor; CTGF, connective tissue
growth factor; MMPs, matrix metalloproteinases; HGF, hepatocyte
growth factor; TSP, thrombospondins; VEGF, vascular endothelial
growth factor; Angio, angiostatin.
MV remodeling correlates with renal (Renal microcirculation,kidney microcirculation)scarring (44), which, in
turn, may subsequently invoke additional changes in vascular
morphology, such as an increase in MV tortuousity (77), which
may reflect the abnormal expansion and development of the renal
vasculature against fibrotic tissue (Fig. 4). Overall, all these
changes may further constrain and limit the already diminished
MV proliferation, growth, and development in the stenotic
kidney, ultimately aggravating MV rarefaction and consequently
renal injury. Hence, timely interventions to preserve a healthy
intact microcirculation(Renal microcirculation,kidney microcirculation) would likely interrupt this injurious feedforward mechanism that precipitates the progression of renal
injury.
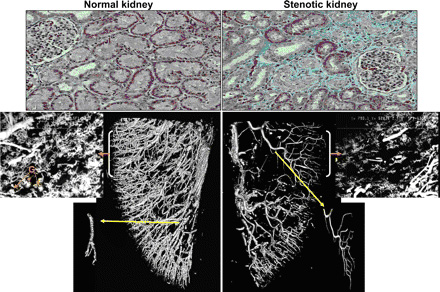
Fig. 4.
Representative picture showing renal fibrosis (top, ˇÁ20) and
three-dimensional micro-computerized tomography reconstruction
of the renal (Renal microcirculation,kidney microcirculation)MV architecture (bottom), tomographically isolated
microvessels (yellow arrow), and cross sections of the
microfilm-perfused kidneys (orange arrow, 250 ¦Ěm, ˇÁ20) in normal
kidneys and in exposed to chronic renal artery stenosis. The
stenotic kidney has a significant MV rarefaction and increased
MV tortuousity. The latter likely reflects an abnormal
development of the intra-renal (Renal microcirculation,kidney microcirculation)vasculature possibly due to the
build-up of renal scarring in the stenotic kidney. G, glomeruli.
Therapeutic Approaches Considering that
mechanisms controlling the generation of new vessels may be
exhausted, negated, or defective during sustained and
progressive renal injury, targeted interventions to preserve the
renal microcirculation (Renal microcirculation,kidney microcirculation)may not only decrease the evolving injury
in renal vascular disease but may also constitute a neoadjuvant
intervention to improve the success of current strategies to
improve renal function, such as revascularization. It has been
shown that frequently used drugs in humans like statins (14, 18,
71), angiotensin (62), and endothelin (9, 39) receptor blockers,
may regulate angiogenesis in different vascular beds, such as in
the heart, brain, and kidney. However, those effects on the
small vessels are reported mainly as collateral rather than as a
main effect. There is a relative lack of targeted interventions
on the renal microcirculation(kidney microcirculation), and in this section, I briefly
discuss promising evidence for such treatments.
Few and small human studies have tested the efficacy of
proangiogenic therapies by direct administration of angiogenic
cytokines (34), but the evidence supporting the administration
of angiogenic factors comes mostly from studies in experimental
settings (1, 33, 48). However, the use of angiogenic modulators
to treat the ischemic kidney is virtually unexplored. The VEGF
pathway has been a target for investigation as a proangiogenic
therapy in ischemic settings (4, 17). We have recently
demonstrated, in proof-of-concept studies, distinct vasculo- and
reno-protective effects of intrarenal administration of VEGF in
the stenotic kidney (7, 37). Although by experimental design,
these effects were largely preventive and at a very early stage
of the disease, the data were promising and support the
necessity of further studies to fully characterize the potential
of such intervention for human disease. Challenges for future
studies and therapeutic intervention are that the onset of RVD
in humans is rarely definable, the patients are diagnosed at
different stages of the disease, and the severity and extent of
renal (Renal microcirculation,kidney microcirculation)compromise varies.
Significant attention has been directed to the biological and
therapeutic capabilities of progenitor cells, an emerging field
of research to treat ischemic tissues. The vascular endothelium
is constantly exposed to mechanical and chemical insults, and
the continuous process of repair is mediated, in part, by
adjacent cells or by recruited progenitors. Circulating cells
play a critical role in healing the endothelium when the
intrinsic system is unable to adequately support tissue repair.
Endothelial progenitor cells mobilized in response to ischemia
play a crucial role in augmenting neovascularization of ischemic
tissues and endothelial replacement after vascular injury.
Defects in the number and/or function of endogenous cell
progenitors have been observed in patients with coronary artery
disease and diabetic nephropathy (25, 53, 59). Furthermore,
clinical and experimental studies indicate that the number and
function of circulating progenitors are also decreased in
chronic renal (Renal microcirculation,kidney microcirculation)disease (31, 51).
Recent evidence suggests potential for cell-based repair in the
acutely injured kidney to augment local regeneration, and this
therapeutic approach to treat the kidney is gaining momentum. We
have recently shown that the chronically stenotic kidney has a
defective repair response to ischemia, since cell progenitors
and the kidney showed abnormal expression of homing
cell-recruitment factors, hence, resulting in abnormal
angiogenesis and MV rarefaction (16). Although retention of
cells in the renal (Renal microcirculation,kidney microcirculation)tissue is low, the administration of cell
progenitors likely leads to autocrine and paracrine effects on
the surrounding cells. These cells reduce renal damage via
secretion of angiogenic growth factors that induce mobilization
of endogenous progenitor cells, which can then migrate and
differentiate into mature vascular endothelial cells, promoting
angiogenesis. In other words, this approach recuperates a renal
(Renal microcirculation,kidney microcirculation)endogenous vasculo-protective mechanism, since intrarenal
administration of autologous endothelial progenitor cells
significantly reversed MV rarefaction in the stenotic kidney,
attenuated renal dysfunction, and decreased fibrosis (13).
Conclusion, Perspectives and Significance
Identifying means to determine the frontier between reversible
and irreversible renal (Renal microcirculation,kidney microcirculation)injury and when renal function may still
be salvageable would have significant clinical impact. Future
studies concentrating on noninvasive assessment of the renal
architecture, and the response to established and novel
treatment options should help on achieving such goals. The
damage of the renal (Renal microcirculation,kidney microcirculation)MV architecture and deterioration of the angiogenic response may constitute early crucial steps in the
complex multiple pathways involved in progressive renal injury,
and as the damage of the renal (Renal microcirculation,kidney microcirculation)parenchyma progresses, the injury
advances toward irreversibility. We have shown that a decreased
cortical MV density in the chronic RVD kidney is associated with
decreased renal blood flow, glomerular filtration rate,
perfusion, and tubular function (10, 13, 14, 36). Such decreases
in the microvasculature of the stenotic kidney affect interlobar,
arcuate, and interlobular arteries, and smaller branching order
microvessels like arterioles, capillaries, and venules. These
deleterious changes in the MV architecture and function
initially compromise the renal (Renal microcirculation,kidney microcirculation)cortex but also extend at the
later stages to the medulla (10, 36), as kidney disease evolves
in our model. Our previous studies have also demonstrated a
substantial deterioration of mainly proximal tubular and Henle's
loop function, as well as tubular atrophy (10) accompanying MV
dysfunction, damage, and loss, indicating a vascular-tubular
damage correlation (3). It is important to emphasize that
experimental evidence from our laboratory supports the notion
that the extent of the overall renal functional and structural
damage could partly be reversible by targeted interventions that
protect the renal microvasculature (13, 37). Do nephrons grow
back or generate by such interventions? Likely not, but it is
possible that restoration of blood flow by generation of new
vessels that shunt preexisting damaged ones and MV repair that
is led by such interventions also contributes to restore
filtration function in partly damaged or hibernated (19, 69) but
potentially recoverable nephrons.
An intact and healthy microcirculation(Renal microcirculation,kidney microcirculation) is vital to restore blood
flow to the injured tissues to initiate a successful repair
response. Modifications in the renal microcirculation
(kidney microcirculation)and
generation of new vessels in the kidney seem to be processes
activated in response to injury to preserve renal function, but
a deregulated or insufficient formation of new vessels could be
deleterious (9, 13, 14, 76). It is possible that, as glomerulosclerosis and tubulo-interstitial fibrosis represent
the final stage of CKD, the deterioration of MV function, MV
damage, and later MV loss may indeed represent the initial steps
of renal (Renal microcirculation,kidney microcirculation)injury in RVD. The resulting tissue ischemia thus
serves as a stimulus for the activation of proinflammatory and
profibrotic factors, leading to renal parenchymal injury and
initiating a potential vicious circle that results in
progressive, irreversible renal injury (Fig. 5). Hence, it is
possible that current failures of therapeutic interventions in
chronic RVD might be partly due to an overlooked or
underestimated severity of renal MV disease at the moment of
treatment.
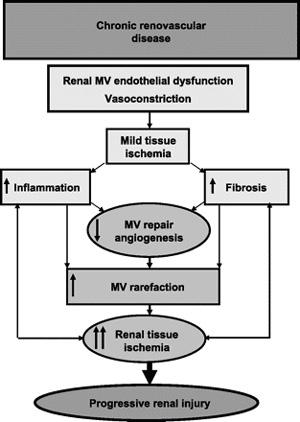 Fig.
5. Fig.
5.
Schematic illustration summarizing the potential role and
mechanisms of MV disease in renovascular disease. Initial renal
(Renal microcirculation,kidney microcirculation)MV endothelial dysfunction may lead to a decrease in blood
supply and mild ischemia, initiating inflammation and fibrosis.
These, in turn, further compromise MV repair, and promote MV
damage and loss, suggesting a vicious circle that ultimately
leads to progressive and, likely, irreversible renal injury.
Targeted interventions to enhance endogenous renoprotective
mechanisms, such as cell-based therapy or the use of angiogenic
cytokines have shown promising results in experimental animals
and in some clinical settings. However, carefully designed
prospective experimental and clinical studies are needed to
determine the appropriate utilization of such therapeutic
options, possibly accompanying established interventional
techniques to ultimately improve the outcomes of patients with
chronic renal (Renal microcirculation,kidney microcirculation)disease.
GRANTS
This article was supported by Grants HL-095638 and HL-51971 from
the National Institutes of Health.
Previous SectionNext Section
DISCLOSURES
No conflicts of interest, financial or otherwise, are declared
by the author.
Copyright © 2011 the American Physiological Society
REFERENCES
1.Balzer KM, Pfeiffer T, Rossbach S, Voiculescu A, Modder U,
Godehardt E, Sandmann W. Prospective randomized trial of
operative vs interventional treatment for renal artery ostial
occlusive disease (RAOOD). J Vasc Surg 49: 667¨C674; discussion
674¨C665, 2009.CrossRefMedline3.↵ Beeuwkes R 3rd., Bonventre JV.
Tubular organization and vascular-tubular relations in the dog
kidney. Am J Physiol 229: 695¨C713, 1975.......etc. |

